
WIP: Interfaces to external programs and libraries
The Fleur-phonopy interface
Fleur can be used in conjunction with the phonopy code [www.phonopy.github.io/phonopy/, "First principles phonon calculations in materials science”, Atsushi Togo and Isao Tanaka, Scr. Mater., 108, 1-5 (2015)] to run phonon calculations in a Finite Displacement approach. For this, please pull and build the release version of phonopy [2.9 or greater] and the development version of Fleur respectively, as the necessary adjustments are not yet in the current Fleur release version [Fleur MaX-5.0]. In the following a short recipe for calculating the phonon dispersion of face-centered cubic aluminium is given as a basic example.
The phonon spectrum of fcc aluminium
A Fleur-phonopy calculation starts from a single inpgen file for a perfect unitcell of the material you want to study. For fcc-Al this means a file (titled fleur_inpgen for example) looking something like this:
Aluminium test Fleur
0.0 1.0 1.0 ! a1
1.0 0.0 1.0 ! a2
1.0 1.0 0.0 ! a3
7.656 ! aa
0.5 0.5 0.5 ! scale
1 ! num atoms
13.1 0.0 0.0 0.0
&atom element="al" id=13.1 rmt=2.50 lmax=12 lnonsph=12 /
&exco xctyp='vwn' /
&comp kmax=4.0 gmax=24.01 gmaxxc=24.01 /
&kpt div1=4 div2=4 div3=4 /
&end /
Several adjustments need to be made in comparison to a normal inpgen file:
- The Bravais matrix must be given directly and cannot be specified e.g. by latsys='fcc'. The overhead for calculating the matrix from this tag and a lattice constant alone is not programmed into phonopy but lives in the Fleur input generator. Scales are allowed. The overall scale of the matrix (i.e. the lattice constant) must be in Angstrom.
- The comment '! a1' must be added after the first line of the Bravais matrix and '! num atoms' after the atom count to make the file parseable for phonopy.
- The initial Bravais matrix, the scales and the atom positions must be given as decimal numbers and can (for now) not be expressed as fractions.
- The namelist
&inputis superfluous and should be omitted, as the interface will assume atom positions given in relative coordinates and a bulk setup.
With these adjustments, phonopy can now be executed to generate the necessary supercell input structures by:
phonopy --fleur -c fleur_inpgen -d --dim="2 2 2"
--fleur tells phonopy to use the Fleur interface, -c specifies the name of the file containing the perfect unitcell, -d tells it to generate the necessary displacements needed to construct the force constants/dynamical matrix from supercell calculations and --dim specifies the size of the supercell in each direction. This will generate several files. A phonopy file called phonopy_disp.yml, an input file for the perfect supercell stucture supercell.in and displaced supercell files named supercell-XXX.in, with XXX enumerating the necessary displacements. The supercell files obviously contain different atom counts compared to the original file, however it is important to note that all additional information after the crystal setup is copied over to the supercell input. That means the initial specification off e.g. the k-point grid is directly determined for the supercells in the simple input file.
Best practice would be to now create subfolders for each displaced supercell input. In each of these subfolders (disp-XXX for example), you can then run the Fleur inpur generator with
inpgen -f supercell-XXX.in
to generate the inp.xml, kpts.xml and sym.xml for a calculation. Set itmax to a value that suffices to converge the calculations or run each of them repeatedly until that is the case by invoking Fleur. Once this is done, switch l_f on in each inp.xml and set the f_level tag to at least 0 (more if you want to use the refined force levels described in the last section). Invoke Fleur again in each subfolder to gain a FORCES file for each calculation. Copy them to the directory where phonopy was originally called and name them FORCES-XXX for example. The next step is to call phonopy to collect these forces into a FORCE_SETS file.
phonopy --fleur -f FORCES-001 FORCES-002 ...
From this file, you can then run several phonopy calculations. The simplest example would be calculating the phonon dispersion curves by calling:
phonopy --fleur -c fleur_inpgen --dim="2 2 2" -p --band="1/2 1/2 1/2 0 0 0 1/2 0 1/2"
to find the phonon spectrum along the path L--X. If everything went right, the spectrum should look something like this:
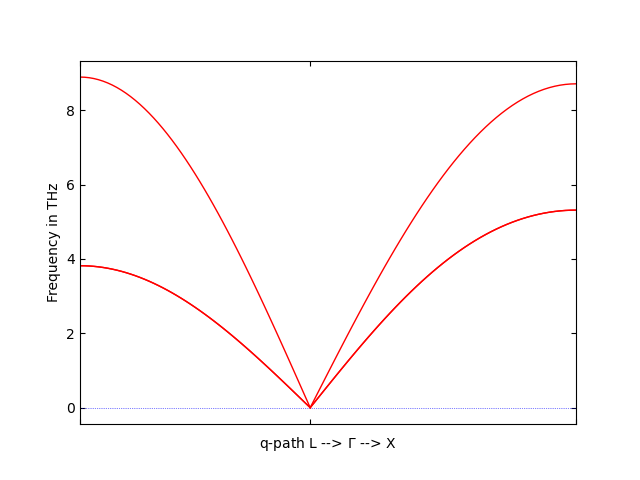
Phonon dispersion in fcc-Al.
This is, of course, the most basic example possible. For further possible calculations consult the phonopy documentation. Should you encounter an error that you are sure is not caused by improper usage of the code, please report it and specifically note that you used the Fleur interface so it is funneled back to the Fleur development team as well.
Using Fleur with the libxc library
Instead of using the exchange-correlation potentials that are found in the Fleur source code, one can also make use of the Fleur interface to the libxc library of functionals [www.tddft.org/programs/libxc/, "Recent developments in libxc — A comprehensive library of functionals for density functional theory", Susi Lehtola and Conrad Steigemann and Micael J.T. Oliveira and Miguel A.L. Marques, SoftwareX, 7, 1-5 (2015)] that provides a broad range of functionals for LDA, GGA, metaGGA and related calculations and their derivatives up to fourth order, drastically reducing the implementation effort of features that use them and instantly increasing the variety of functionals available in a DFT code.
To use a standard LDA/GGA functional, the corresponding tag in the inp.xml needs to be modified:
<xcFunctional name="LibXC" relativisticCorrections="F">
<LibXCName exchange="gga_x_pbe" correlation="gga_c_pbe"/>
</xcFunctional>
The example above would yield a calculation with the GGA functional of Perdew, Burke and Ernzerhof. A comprehensive list of possible correlation and exchange functionals is found at www.tddft.org/programs/libxc/functionals/. The following subsections cover some of the nitty-gritties considering their usage.
GGA functionals and the chain rule
One point to keep in mind is the handling of GGA funtionals in libxc. While for (possibly spin-dependent) LDA calculations, libxc takes only the up- and down densities as arguments and can provide (mixed) derivatives by these densities up to fourth order, there are more arguments in the GGA case.
As additional input, libxc GGA expects the so-called 'conjugated gradients', i.e. the scalar products for and up-up/up-down/down-down corresponding to . It also provides the derivatives with respect to these products and the spin-dependent densities as explained at www.tddft.org/programs/libxc/manual/libxc-5.1.x/) for version 5.1.X. This necessitates some post-processing when used in the Fleur code.
For a spin-dependent density and GGA functional, the exchange-correlation potential is defined as:
But the functional and its derivatives in libxc are provided as instead. This means Fleur has to apply the chain rule for derivatives to get the correct potential. For non-spin-polarized and spin-dependent densities respectively (and directly using the notation of the libxc manual), this looks as follows:
In the MaX-5.0 version of Fleur, comparative calculations show that GGA functionals hard-coded into Fleur and taken from libxc do not yield entirely identical , exactly due to the necessary added calculations of the chain rule. The LDA functionals can, however, be used without further caveats.