
1. Force Relaxations and Thin Films
1.1 Relaxing the atom positions in a single MoS2 layer
MoS2 is a transition metal dichalcogenide that crystallizes in a layered structure in which each layer features a honeycomb lattice with a 3-atomic basis. The interlayer-bonding has a Van-der-Waals nature which translates into a large interlayer distance. Single layers of this material can also be exfoliated to obtain a 2D material. In this exercise we consider such a single MoS2 layer as a thin film system. The following inpgen input describes the unit cell starting point we use for the calculations:
MoS2 monolayer &input film=T symor=t/ 0.5 -0.5 0. 0.5 0.5 0. 0. 0. 1. 5.952636864 1. -3. 30.00 3 42 0.333333 0.666667 0.00 16 0.666667 0.333333 2.85 16 0.666667 0.333333 -2.85 &factor 1. 1. 1. / &end /
The &input namelist here specifies that this is a film system (film=T). We also only want employ symmorphic symmetry operations (symor=t). The latter switch is used because in Fleur non-symmorphic symmetry operations are not compatible to all lattices. If you set up own input files and obtain a Fleur error message about this you now know what to do.
Next comes the Bravais matrix which is scaled according to the two lines following it. The first line is a general scaling parameter for the Bravais matrix, the second line specifies scaling parameters for each column of the matrix separately. Note that a scaling of '-3' actually means a scaling with . The last entry ('30.00') is just a large number which only ensures that the film fits into the here-specified unit cell. It is automatically reduced to a reasonable value by the program.
For film systems the last atom coordinate is always cartesian. This is why we have larger numbers in comparison to the other two coordinates which are provided in terms of Bravais lattice vectors. There are more scaling parameters for the atom positions below the last atom position entry. The atom positions are divided by these numbers. Here these scaling parameters are .
Note that the x and y atom positions are supposed to be at and . We provide these numbers with a precision better than . With such a precision they are automatically turned into the exact values. A less precise specification of the atom positions will lead to a reduced set of symmetry operations being detected. A consequence of this is the generation of a k-point set that breaks the non-detected symmetries resulting, e.g., in the calculaton of inaccurate forces that are not compatible with all symmetries actually present in the system. Of course, such a problem can be overcome by using a very fine k-point mesh, but it is more efficient and precise to make sure that all relevant symmetry operations are included in the calculation. You can also directly provide the exact fractions by using the scaling factor. With this the atom positions would look like:
42 1.0 2.0 0.0 16 2.0 1.0 2.85 16 2.0 1.0 -2.85 &factor 3.0 3.0 1.0 /
The z-position of the sulfur atoms provided in this inpgen input is only a guess. In this exercise we will optimize this position with respect to the forces on the atoms and compare the band structures with and without the relaxation.
Use the inpgen input to generate a Fleur input for the MoS2 monolayer.
The calculation of forces is based on the presence of a self-consistent density for the respective unit cell. Furthermore the movement of atoms requires nonoverlapping MT spheres for the atom configuration after the movement. For this we will first adjust a few parameters in the inp.xml file:
- Set the number of iterations (itmax) to 50. The convergence here is not problematic. Therefore this just leads to the convergence criterion ending the calculation.
- Multiply the MT radii for the atoms by .
If the respective switch is on forces are calculated once the density convergence criterion is met.
Run Fleur to reach self-consistency. After this has succeeded copy the files to two subdirectories 'bands' and 'relax'.
Perform a band structure calculation in the 'bands' folder. The resulting band structure should look similar to the following plot.
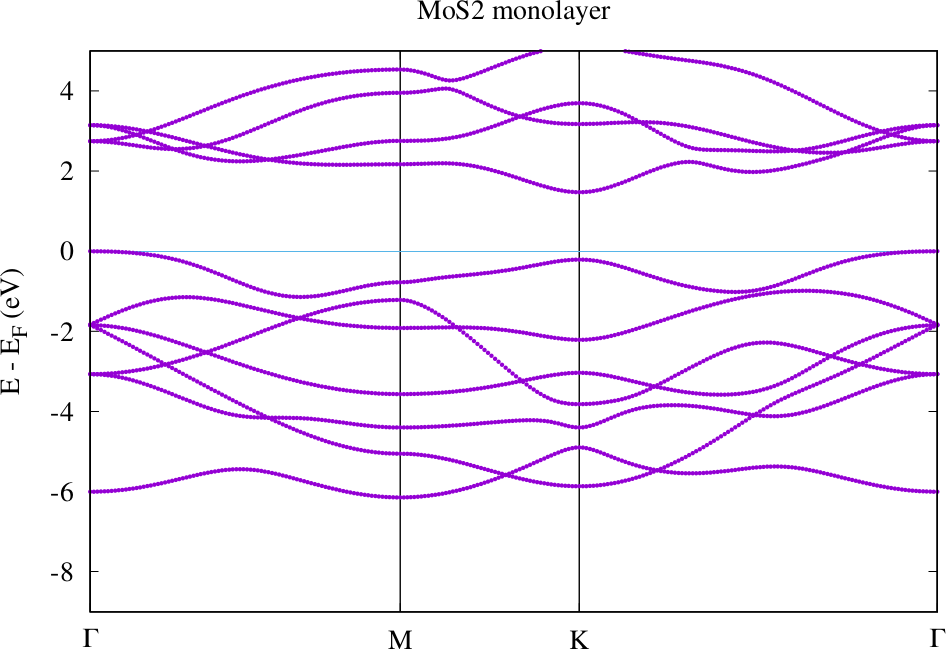
In the 'relax' folder we now modify the inp.xml file to perform the force calculations and generate the atom displacements. For this set the /fleurInput/calculationSetup/geometryOptimization/@l_f switch to "T".
In the geometryOptimization XML element you can also change the relaxation algorithm to something different (we keep BFGS), change the mixing parameter, or adjust the criteria for a successful relaxation. Note that you can limit the force calculations for each atom type to certain orientations. This is done in the /fleurInput/atomGroups/atomGroup/force/@relaxXYZ XML attribute. Each logical in this variable stands for one direction. But for this exercise we don't use this switch.
Invoke Fleur to calculate the forces for this configuration.
You will obtain a file 'relax.xml'. In every Fleur run this file is automatically included in the inp.xml file whenever it is present. Investigate the file. You will first find a list of displacements for each atom group. This is used to modify the atom positions in the inp.xml file in Fleur calculations. The displacements are updated by every new force calculation. The other big block in the relax.xml file is the relaxation history. Here you find for every relaxation step and atom type a list of atom positions in cartesian coordinates followed by the forces on the representative atom of this atom type in Hartree per . You can use this list as an output, e.g., look up the final atom positions in it, but it is also an input for Fleur that is used by the relaxation algorithm to determine the next atom positions. An experienced user may modify this file whenever a relaxation process seems to be problematic. For example in certain situations it may be a good idea to delete a part of the relaxation history.
We now have to repeat the self-consistency calculation followed by the force step several times to reach the equilibrium atom positions. You can just invoke Fleur several times to do so. Have a look at the changes in the relax.xml file while you do so. Note that a systematic approach to an automatization of this workflow would be possible by using Aiida. Note also that the Fleur version we are using for this tutorial is compiled with HDF5 support. Without HDF5 support the output files would be slightly different and the user would have to delete the 'stars' and 'wkf' files after every force calculation step. These files depend on the atom positions.
If you don't want to manually invoke Fleur repeatedly you may write a tiny script 'runscript.sh' that does this a few times:
Fleur Fleur Fleur Fleur Fleur Fleur Fleur
Replace in the script 'Fleur' by the Fleur call you perform on your system. Make the script executable by running the command 'chmod a+x runScript.sh'.
Once the atoms have reached their equilibrium positions find the sulfur z-positions of the last force step in the relax.xml file and compare them to the original positions. How much did they change? What is the energy difference between the two configurations? Your relax.xml file should look similar to the following output from the reference calculation (modified by removing some irrelevent digits):
<!-- Attention, absolute coordinates used here -->
<relaxation>
<displacements>
<displace> 0.0000000000 -0.0000000001 0.0000000000 </displace>
<displace> 0.0000000000 0.0000000001 0.1255317452 </displace>
</displacements>
<relaxation-history>
<step energy=" -4848.0786605687">
<posforce> 0.0 -3.4367564958 0.0000000000 0.0 0.0 0.0000000000 </posforce>
<posforce> 0.0 3.4367564958 2.8500000000 0.0 0.0 0.0262595326 </posforce>
</step>
<step energy=" -4848.0799114622">
<posforce> 0.0 -3.4367564958 0.0000000000 0.0 0.0 0.0000000000 </posforce>
<posforce> 0.0 3.4367564958 2.8762595326 0.0 0.0 0.0203653455 </posforce>
</step>
<step energy=" -4848.0819723607">
<posforce> 0.0 -3.4367564959 0.0000000000 0.0 0.0 0.0000000000 </posforce>
<posforce> 0.0 3.4367564959 2.9669903572 0.0 0.0 0.0016385907 </posforce>
</step>
<step energy=" -4848.0819943703">
<posforce> 0.0 -3.4367564959 0.0000000000 0.0 0.0 0.0000000000 </posforce>
<posforce> 0.0 3.4367564959 2.9749293025 0.0 0.0 0.0001148937 </posforce>
</step>
<step energy=" -4848.0819950663">
<posforce> 0.0 -3.4367564959 0.0000000000 0.0 0.0 0.0000000000 </posforce>
<posforce> 0.0 3.4367564959 2.9755279353 0.0 0.0 0.0000007598 </posforce>
</step>
<step energy=" -4848.0819950697">
<posforce> 0.0 -3.4367564959 0.0000000000 0.0 0.0 0.0000000000 </posforce>
<posforce> 0.0 3.4367564959 2.9755319205 0.0 0.0 -0.0000000332 </posforce>
</step>
<step energy=" -4848.0819950695">
<posforce> 0.0 -3.4367564959 0.0000000000 0.0 0.0 0.0000000000 </posforce>
<posforce> 0.0 3.4367564959 2.9755317534 0.0 0.0 -0.0000000016 </posforce>
</step>
<step energy=" -4848.0819950696">
<posforce> 0.0 -3.4367564959 0.0000000000 0.0 0.0 0.0000000000 </posforce>
<posforce> 0.0 3.4367564959 2.9755317452 0.0 0.0 0.0000000000 </posforce>
</step>
</relaxation-history>
</relaxation>
Note: The cdn.hdf file has become quite large because many densities are stored in it. You can investigate it by calling fleur with the "-info" command line switch. You will obtain a list of the densities together with time stamps and the distance to the previous density. Of course, there are ways to get rid of certain densities. One way is to perform the Fleur calculations with the command line switch "-last_extra". This will write another file out in every iteration that only contains the last charge density. Another way is to use Fleur command line options to actually delete densities from the cdn.hdf file. A list of all Fleur command line options can be obtained with the command line option "-h". But in this exercise we will not manipulate the cdn.hdf file in such a way.
Create a subdirectory 'bands' and copy all files into it. Switch of the l_f switch and calculate the band structure. The result should look similar to the following plot.
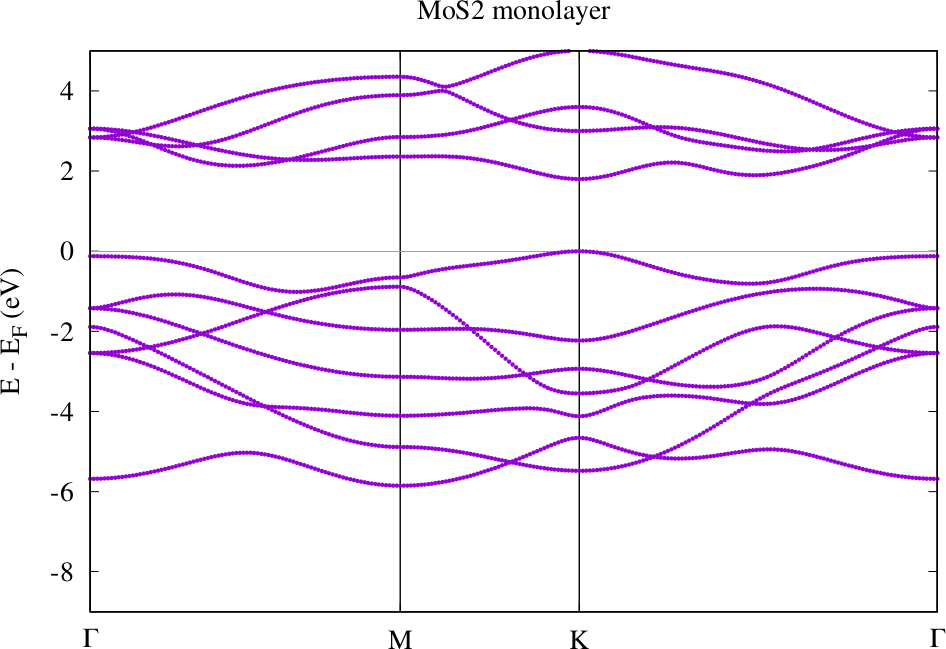
Compare the band structures for the guessed atom positions and the relaxed atom positions. Where are the valence band maxima and conduction band minima in each case? How large are the band gaps for each configuration. If everything worked correctly you can observe that the small change in the atom position has a rather large effect on these quantities.
For a Kohn-Sham system the eigenenergy of the highest occupied state has a strict physical meaning. In Fleur the definition of the Fermi energy at 0K (grep Fermi out.xml) coincides with this quantity. For thin films we can relate this to the vacuum level at an infinite distance from the surface (grep vzInf out.xml) to calculate the work function for a material. What is the value for the work function of MoS2?
Sidenote: Thin film calculations can be used to calculate the properties of surfaces of bulk systems. For this the film thickness has to be increased until the quantities of interest reach stable values. Depending on the material one may either need only a few atomic layers of even more than 20 or 30.
2. Exercises
2.1. PbTiO3 (1)
Lead titanate (PbTiO3) is a ferroelectric material that forms a perovskite structure. Materials in perovskite structure have the chemical formular ABX3. In its basic form perovskites are related to a cubic unit cell with "A" atoms at the corners, "B" atoms in the center, and "X" atoms at the face centers. Look up a visualization of perovskite unit cells. Note that these visualizations are sometimes shifted such that the "B" atom is at a corner position of the unit cell.
As a ferroelectric material PbTiO3 features a spontaneous electric polarization. This causes a slight deviation of the actual unit cell from the basic perovskite structure. In this exercise we want to determine this unit cell under the (wrong) assumption that it is still cubic. We start with the following inpgen input:
PbTiO3
&lattice latsys='sc' a=7.50 /
5
82 0.00 0.00 0.000
22 0.50 0.50 0.475
8 0.50 0.50 0.000
8 0.50 0.00 0.500
8 0.00 0.50 0.500
&end /
As you can see the Titanium atom in this input is slightly displaced from its center position. This is done to prevent inpgen from detecting certain symmetries in the unit cell that would become a problem when we displace atoms in it in z direction.
After generating the inp.xml file place the Ti atom in the center of the unit cell again. To allow for a wider range of atom positions reduce the MT radii of Pb and Ti to 90% and those of O to 95%.
We now perform several self-consistent density calculations for different displacements of the Ti atom in z direction. Vary the Ti position from 1.00/2.00 to 1.05/2.00 in steps of 0.01/2.00 (or finer if you want to have a nicer result, e.g., 0.005/2.00). For each of the displacements calculate the forces on the Ti atom in z direction.
NOTE: If all forces vanish (e.g. due to symmetries) at the moment you can not start with l_f="T" without first having performed a few SCF iterations. The convergence criterion seems to be broken here such that this would lead to a direct calculation of the forces without first obtaining a self-consistent density. Be cautious about this.
Plot the total energy vs. the displacements on top of a plot of the forces vs. the displacements. If everything works as expected you should see a total energy minimum for a finite but small displacement of the Ti atom. The total energy minimum should agree with the vanishing of the force on Ti.
Results to be delivered: Plots with total energy vs. Ti atom displacement and forces on Ti vs. Ti atom displacement.
2.2. PbTiO3 (2)
Starting from a position of the Ti atom near the energy minimum perform a force relaxation for the Titanium and Oxygen atoms in z direction (use the relaxXYZ flag to keep Pb fixed at the predefined position). If you encounter any errors or unexpected behavior in this process consider the hints below. Where are the atom positions for the relaxed unit cell? What total energy does the unit cell feature? Compare the displacements and total energy to the basic perovskite structure and the equilibrium position in the displacement of the Ti atom alone.
Note: In the test calculations to this exercise about 20-30 force relaxation steps were needed to reach the "official" message that the structure is relaxed with respect to the forces:
*****************************************
Run finished successfully
Stop message:
Structural relaxation: Done
*****************************************
This message is reached when in one step of the calculation the inp.xml file values calculationSetup/geometryOptimization/@epsdisp for the atom displacement and calculationSetup/geometryOptimization/@epsforce for the forces on the atoms are underrun. (They were kept at the default values for the test calculation.) The values for the forces are always provided in .
Note that the required steps for structural relaxations may strongly depend on the quality of the initial atom positions.
Results to be delivered: Positions of all (representative) atoms after the force relaxation. Difference in total energies between unrelaxed and relaxed structure.
3. Last weeks results
4. Hints in the context of force relaxations
Most problems in force relaxations are due to one of the following two reasons:
- It may be that in one step atoms are displaced by a far distance. In such a case the last density stored in the cdn.hdf file might be a very bad starting point resulting in a messed up potential and errors due to this. Very large distances in the first self-consistency iterations after such a displacement may also be seen due to this cause. Deleting the 'cdn.hdf' file is one approach to deal with this.
- Typical optimization algorithms are based on the assumption that the function E (e.g. the energy) to be optimized features a parabolic
dependency on its parameters (e.g. atom positions) near its minimum. It is also assumed that the initial guess for the parameters is in a
region where this is the case. If these conditions are not met the next displacements may be very bad or an error might occur, e.g., you
might see some message like 'bfgs: