
1. Convergence with respect to parameters
There are several parameters that control the precision of DFT calculation. Some of them depend on the concrete approach used to implement DFT, others are relevant for all approaches. In this tutorial we want to systematically converge our results with respect to these parameters.
1.1. fcc Cu and Kmax
The LAPW basis set size is mainly controlled by . This is a reciprocal plane-wave cutoff parameter used to specify which plane waves are used to represent the wave functions in the interstitial region of the unit cell. Similar parameters are also present in plane-wave pseudopotential or projector-augmented-wave codes. In some codes this parameter is specified in terms of an energy. Assume the kinetic energy operator (here in Hartree atomic units). What kinetic energy does a plane wave with a of have (in Hartree and in Rydberg)?
In Fleur we also have the parameters and which are used to limit the expansion of the density and the XC potential in terms of plane waves. A condition the three plane-wave cutoff parameters have to fulfill is .
We want to test for one of our test systems (choose fcc Cu with a lattice constant near the equilibrium lattice constant) the dependence of the total energy on . For this we perform self-convergent total energy calculations with increasing in steps of starting with the default until the total energy seems to be stable. Make sure to set fitting (and same) , for all calculations (assume that over the calculations you might increase by up to ). How large is the total energy difference between the calculations with the default parameters and the one with the largest . Plot ((total energy)-(min(total energies))) vs. .
Note: The cutoffs can be set in the XML element calculationSetup/cutoffs.
Note2: The basis set size depends cubically on . On the other hand for large unit cells the algorithm scales cubically with the number of basis functions. This implies an asymptotic scaling of the runtime with . Fortunately does not depend on the unit cell size. It does, however, vary from material to material.
Your results should be similar to what is shown in the following figure.
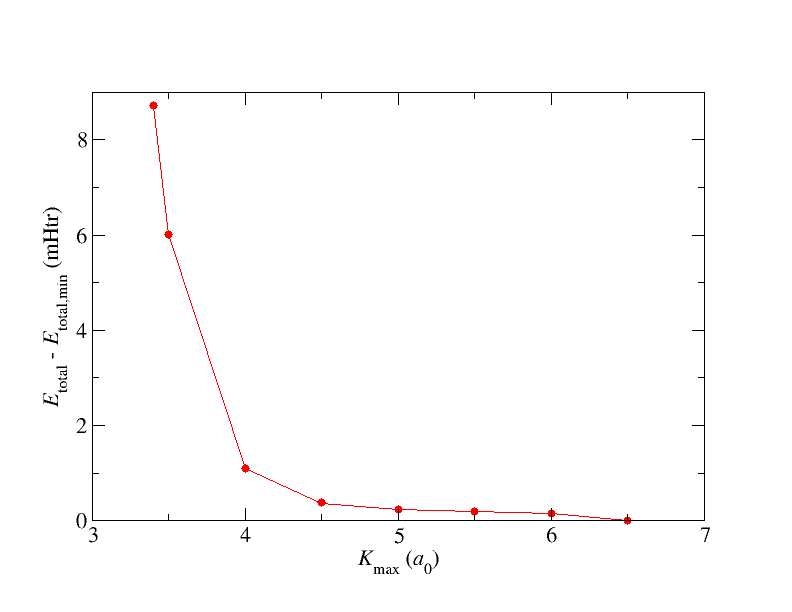
When increasing the total energy should decrease and stabilize at some point. The observable dip for very high is due the fact that the LAPW basis functions are not orthogonal to each other. This implies that at some point the LAPW basis becomes linearly dependent. The dip is a numerical consequence of this. For even larger the calculation would fail with an error. A reasonable choice for is within the most stable region or at least in a region where the quantities of interest do not depend on within the desired precision. Note that reducing MT radii increases the range of the stable region.
1.2. lmax and lnonsph
For the calculation with the converged Kmax we now converge the lmax and lnonsph cutoffs for each atom (if you have multiple atom species in principle these are separate optimizations but in such a case we perform the optimizations simultaneously). The lmax cutoff parameters limit the spherical harmonics expansion of the basis functions in the MT spheres. Beyond this cutoff the basis functions are discontinuous at the respective MT boundary. As such discontinuities are related to large kinetic energy contributions care has to be taken to obtain reasonably smooth basis functions, even if physical effects are related to smaller angular momentum quantum numbers. A rule of thumb for the choice of lmax is , where is the respective MT radius.
Simultaneously with lmax we can also increase lnonsph, though the only connection of this parameter to lmax is . lnonsph has a stronger connection to the actual physics of the material under investigation as it determines up to which of the basis functions the contributions originating from the nonspherical part of the potential in the MT sphere are considered. Typically we choose . For many materials lnonsph can be choosen smaller than lmax but for certain calculations it may be more reasonable to have . Note that increasing lnonsph is computationally much more expensive than increasing lmax ( vs. ).
We perform total energy calculations for increasing lmax, lnonsph values in steps of 2, starting with the default value. Plot ((total energy)-(min(total energies))) vs. .
Your results should be similar to what is shown in the following figure.
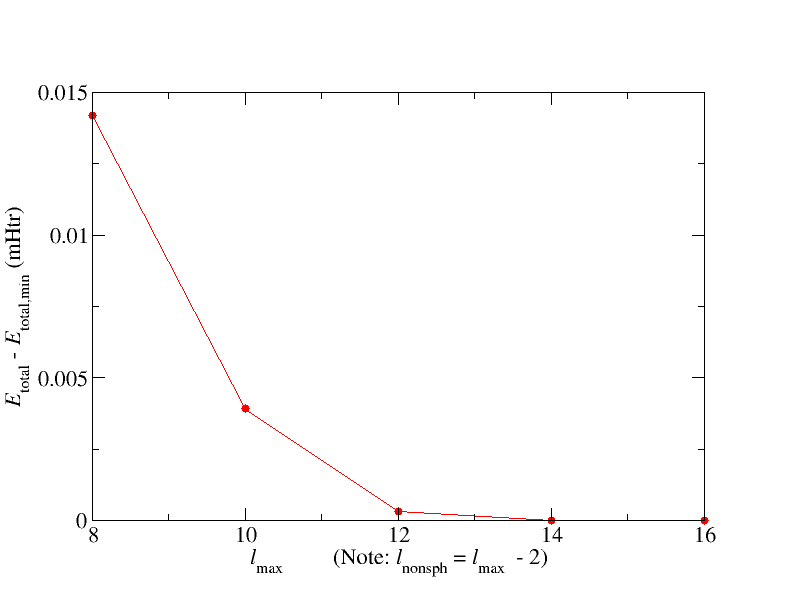
The dependence of the total energy on is rather small here. Please note that this is not a general observation. For other materials may be a more critical parameter.
1.3. k point set and Fermi smearing
All electronic structure calculations for crystals require a k point sampling of the reciprocal unit cell. This is primarily needed for the Brillouin zone integration required for the construction of the electron density. Similarly to the previous two tasks we test the convergence of the total energy with respect to the k point set. For this we increase the k point sampling in each direction simultaneously in steps of 3 until the result is stable, starting with an mesh. Plot ((total energy)-(min(total energies))) vs. numkPointsInOneDirection.
The following plot is an example of what may be observed (taken from other calculations on the same system, there may be some differences). Note that you probably don't want to employ the larger -point sets used for those calculations (this is also not required for this exercise).
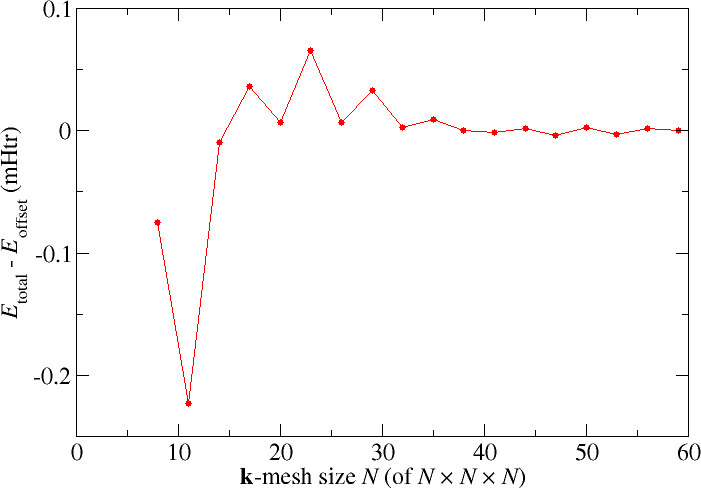
Note that the k point sampling is more demanding for metals than it is for insulators. This is due to the complexity of the electronic structure near the Fermi energy in combination with the occupation of states. For materials with a very complex electronic structure around the Fermi energy a too small k point sampling may even impede the convergence of the DFT calculation, i.e., the iterative procedure does not find a self-consistent solution. One reason for this can be the shifting of states around the Fermi energy from iteration to iteration. This can lead to large changes in the occupation of states. To soften such effects one typically introduces a smooth occupation function. Often this is a Fermi-Dirac distribution, parametrized by an energy or temperature. In Fleur we provide the fermiSmearingEnergy in calculationSetup/bzIntegration/@fermiSmearingEnergy (in Hartree). Alternatively we can also provide a temperature to specify the smearing.
After we reached a self-consistent solution with optimized Kmax, lmax, k point set we reduce the fermiSmearing by a factor of 10 starting with the self-consistent result for the default smearing. How large is the energy difference?
2. Excercises
NOTE: REFERENCE RESULTS FOR LAST WEEKS EXERCISES CAN BE FOUND BELOW. THEY MAY BE NEEDED TO PERFORM THIS WEEKS EXERCISES.
2.1. Parameter convergence for hcp Cu
As practiced above we also converge the parameters for hcp Cu.
Note that our structural optimization kept the c/a ratio fixed. Of course, this is another degree of freedom that can be optimized, but we ignore it here.
Results to be delivered: Plots of the convergence curves for total energy vs. , , , -point mesh; numbers for total energy vs. Fermi smearing energy.
2.2 fcc vs. hcp Cu revisited
Decide on a common set of optimized parameters for the fcc and hcp calculations. Which criteria do you use for that decision? Optimize the lattice parameters and perform Birch-Murnaghan fits. How large are the deviations from the previous results. To what extent is the energy difference between fcc and hcp Cu affected by these more sophisticated calculations. Compare between the calculations with default parameters and those with optimized parameters the total energy difference and relate this to the changes in the difference of the fcc vs. hcp structures.
Results to be delivered: Plots with DFT results and Birch-Murnaghan fits for calculations with optimized parameters, the results for the requested quantities in the paragraph above.
3. Last weeks results
3.1. Cu
- Note: Results may slightly depend on the choice of the MT radii. For the following calculations was chosen.
- reference fcc lattice constant:
- fcc equilibrium lattice constant:
- fcc bulk modulus:
- fcc total energy minimum (1 atom in unit cell):
-
fcc Cu inpgen input:
Cu bulk fcc structure
&lattice latsys='cF' a=6.82 /
1 29 0.0 0.0 0.0
&kpt div1=14 div2=14 div3=14 /
-
Calculation of reference hcp lattice parameter a:
- hcp equilibrium lattice parameter
- c/a is kept constant (optimal value for close packing of spheres)
- hcp bulk modulus:
- hcp total energy minimum (2 atoms in unit cell):
-
hcp Cu inpgen input:
Cu bulk hcp structure &input cartesian=f / &lattice latsys='hcp' a=4.82247 c=7.87506 / 2 29 0.0 0.0 0.0 29 0.6666666667 0.3333333333 0.5
&kpt div1=14 div2=14 div3=7 /
-
hcp - fcc energy difference per atom:
3.2. Ar
- equilibrium lattice constant:
- bulk modulus:
- total energy minimum:
-
inpgen input:
Ar bulk
&lattice latsys='cF' a=9.94 /
1 18 0.0 0.0 0.0