
Partitioning of the unit cell and energy ranges
The setup of unit cells typically starts with the specification of the shape of the unit cell. This is done by defining a Bravais matrix containing the lattice vectors. For many unit cells the entries in the Bravais matrix are not independent of each other because the shape of the cell features certain symmetries. According to these symmetries Bravais matrices can be classified into certain crystal families and lattice systems, each specified by a reduced set of parameters.
In the Fleur input file Bravais matrices are defined in the cell/bulkLattice section for bulk systems and in cell/filmLattice
for thin film systems. Considering bulk systems they can either be defined explicitely (if cell/bulkLattice/@latnam="any") or by providing
the name of the lattice system in cell/bulkLattice/@latnam. In the explicit case the Bravais matrix is then provided in cell/bulkLattice/BravaisMatrix,
if the lattice system is specified only the respecive parameters have to be provided (cell/bulkLattice/a1, cell/bulkLattice/a2, cell/bulkLattice/c).
The FLAPW method is based on a partitioning of the unit cell into different regions. As sketched in figure 1 it is divided into non-overlapping so-called muffin-tin (MT) spheres centered at each atomic nucleus and an interstitial region (IR) between the spheres.
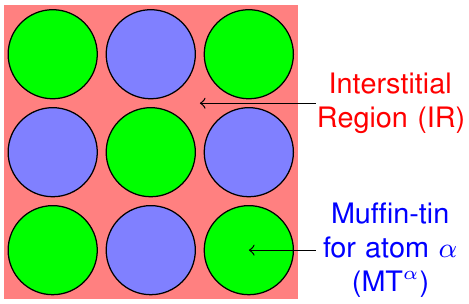
Partitioning of the unit cell in non-overlapping MT spheres and an interstitial region.
The representation of the Kohn-Sham eigenstates as well as the density and the potential varies between these regions and can be adjusted by cutoff and other numerical parameters for each region.
In the input file for each chemical element present in the unit cell one or more species (atoms with same numerical parameters) are
defined in atomSpecies. For each species many parameters are defined. This includes an identifier (atomSpecies/species/@name)
and the atomic number (atomSpecies/species/@atomicNumber).
The name of the chemical element is also shown (not an input parameter) (atomSpecies/species/@element).
The respecive MT radius is set in atomSpecies/species/mtSphere/@radius.
Atoms from the same species are grouped according to their symmetrical equivalence in the unit cell (atomGroups).
Each atomGroups/atomGroup is connected to a species via the respective identifier (atomGroups/atomGroup/@species).
The atom positions related to each atom of an atom group are specified in different ways within atomGroups/atomGroup.
atomGroups/atomGroup/relPos specifies a position in units of lattice vectors. In atomGroups/atomGroup/filmPos an atom position
is specified by lattice vectors in the xy plane and an absolute coordinate in z direction.**
Another subdivision is performed with respect to the energy levels of the electrons in the unit cell. Figure 2 sketches the different classes of electron states obtained by this procedure.
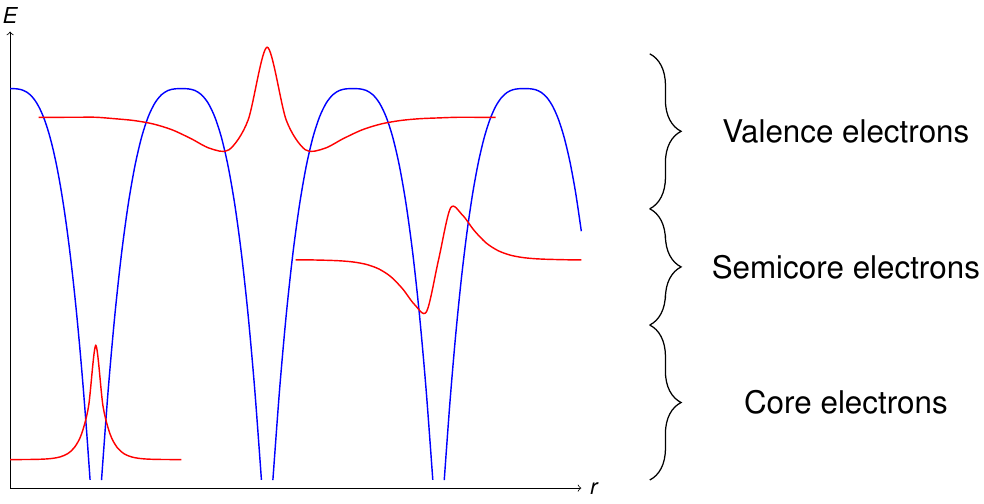
Classification of the electrons according to their energy levels. The blue curve is the potential.
Energetically deep lying electrons are denoted as core electrons. These electrons are localized within the MT sphere of the respective atom and should be described fully-relativistically.
On the other hand electrons near the Fermi energy are considered to be valence electrons. They are typically delocalized and in crystals with periodicity have to be described as Bloch states
with
Energetically between the valence and the core electrons there may be electrons which can neither be considered to be fully localized within the respective MT sphere, nor delocalized.
In FLAPW the concrete classification of the electrons has to be performed by the user as an input to the calculation. This is typically supported by reasonable default classifications. However, these default classifications may not work for every unit cell since the size of MT spheres depends on bond lengths and may vary strongly for different chemical compositions.