
Calculation a Bandstructure
Starting with FLEUR version .24 (used for the NIC winter school 2006) there is a convenient new run mode that further simplifies the creation of band structure plots.
For a proper band structure plot, also the correct Fermi energy is desired. This leads to two options:
The quick way.
In this way no special care is taken for the Fermi energy, it is obtained from the eigenvalues of our band structure. Due to the very selection of the k-points, this can deviate from the true Fermi energy you obtained in your self-consistency. In the case of semiconductors, your band structure as well as your self-consistency k-point set might contain the k-point comprising the highest occupied state, then there should not be any difference. In the case of metals, one will expect a larger shift. The new mode tries to guess a reasonable path in the Brillouine zone for the k-points. For many simple cases it will give a suitable "default bandstructure". However, by supplying a band_inp file you can also choose the k-points yourself.
In the new mode you tell FLEUR to create a new k-point set and calculate the eigenvalues. Therefore you either need to remove all k-point dependent files,
> rm broyd* fl7para kpts pot* wkf2
or you continue the calculation in a new directory:
> mkdir band && cp inp cdn1 enpara sym.out band && cd band
As a next step, you modify the switch band in the inp file, changing the line from
iplot=F,score=F,plpot=F,band=F
to
iplot=F,score=F,plpot=F,band=T
That's it, now you can run FLEUR,
> fleur.x
DOS OK
and see newly created files bands.1 and band.gnu. The latter one is a GNUplot script that can be used to easily create a postscript file: (Note that you have to adjust the Fermi-energy as showed below.
> gnuplot < band.gnu > band.ps
that can be viewed e.g. by
> gv band.ps
For the Silicon setup used in this guide, it looks like this:
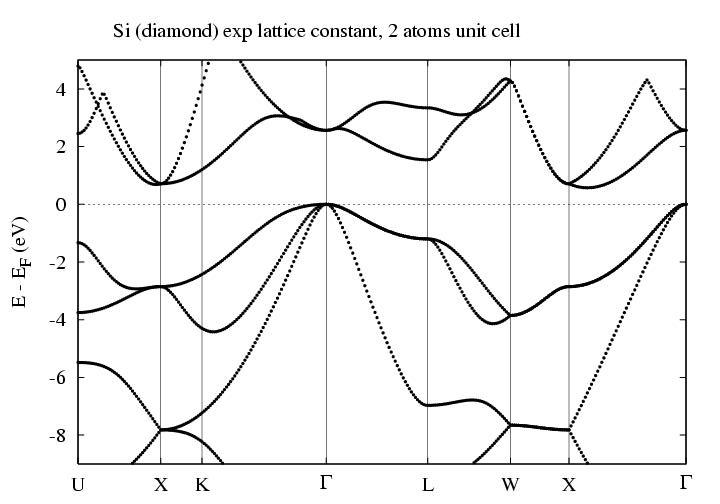
By the way, if you watch your kpts file,
> head -1 kpts
301 1.0000000000 you see that the number of k-points used is 301, since nkpt= 300 was specified in the inp file.
The proper way.
In the case of silicon, the Gamma point (comprising the largest occupied eigenvalue) is contained in our band structure path but not in the Monkhorst pack set used for self-consistency, therefore we expect a small change in the Fermi energy. To make matter clearer, the numbers for the Fermi energy below are taken from a cupper bulk calculation. Nevertheless, the procedure itself is general.
First remember the correct Fermi energy, so do
> grep -i Fermi out
fermi energy and band-weighting factors: FERMIE: FERHIS: Fermi-Energy by histogram:
EF_NEWTON: Adjust Fermi-Energy by Newton-Method.
--> new fermi energy : 0.281417 htr
after your self-consistency (This is only relevant for metals, for insulators or semiconductors the Fermi level is not printed explicitely in the out file). Then remove only the two files,
> rm fl7para kpts
and modify the switches dos and ndir in the first line of the inp file (same as above) as well as the pot8 switch, changing it from
strho=F,film=F,dos=F,isec1=99,ndir= 0,secvar=F
:
:
vchk=F,cdinf=F,pot8=F,gw=0,numbands= 0
:
:
iplot=F,score=F,plpot=F,band=F to
strho=F,film=F,dos=T,isec1=99,ndir= 1,secvar=F
:
:
vchk=F,cdinf=F,pot8=T,gw=0,numbands= 0
:
:
iplot=F,score=F,plpot=F,band=T Now run FLEUR again
> fleur.x
DOS OK and grep for the new Fermi energy:
> grep -i Fermi out
FERMIE: FERHIS: Fermi-Energy by histogram:
EF_NEWTON: Adjust Fermi-Energy by Newton-Method.
--> new fermi energy : 0.271041 htr
There is a difference of 0.281417 htr - 0.271041 htr = 0.010376 htr = 0.282227 eV that the correct Fermi energy is higher, i.e. with respect to the Fermi level the newly calculated eigenvalues should be lowered by this number. For this, edit the last line of the GNUplot script band.gnu from
"< awk '{print $1,$2+0.00}' bands.1" w p pt 7 ps 0.5
to
"< awk '{print $1,$2-0.282}' bands.1" w p pt 7 ps 0.5
Now you can continue as above,
> gnuplot < band.gnu > band.ps && gv band.ps