
More advanced Bandstructure calculation
This tutorial uses results of previous tutorial
Please first run the calculations of:
and then copy the results here.
cp -r ../F6/MoS2 .; cd MoS2/relax
Create a subdirectory bands, copy all files into it and change to the new directory:
mkdir bands ; cp * bands ; cd bands
The band structure with the relaxed structure and orbital projections
We start by changing the inp.xml file again.
Switch off the l_f switch and calculate the band structure by first generating the desired k-point set:
inpgen -inp.xml -kpt band=60
Afterwards edit the inp.xml file as sketched for the nonrelaxed case and invoke fleur:
fleur_MPI
For the "path-2" k-point path the result should look similar to the following plot.
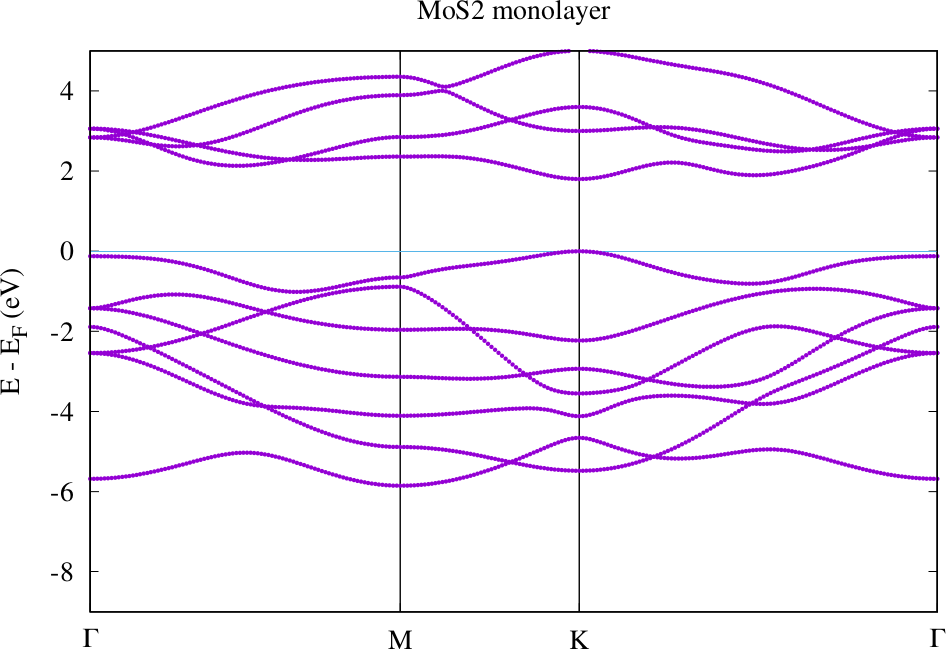
Band structure for MoS2 monolayer with geometry after force relaxation.
Compare the band structures for the guessed atom positions and the relaxed atom positions. Where are the valence band maxima and conduction band minima in each case? How large are the band gaps for each configuration (grep bandgap out.xml). If everything worked correctly you can observe that the small change in the atom position has a rather large effect on these quantities.
grep bandgap ../out.xml
grep bandgap ../../out.xml