
The standard self-consistent field (SCF) calculation
Directly after running the Input generator inpgen the Fleur input file is set up such that
Fleur will perform an SCF calculation with it.
The calculation of a self-consistent density starts with the construction of a starting density which is automatically generated if no other density is present in the working directory. It is a superposition of atomic densities generated on the fly. If there already is a working density in the calculation directory Fleur starts the calculation with this density.
If Fleur is linked to the HDF5 library charge densities are stored in the file cdn.hdf.
Without HDF5 the working density is stored in the file cdn1 and previous densities are
stored in the files cdn?? where the question marks stand for two digits following the
cdn. Note that the working densities should be consistent with the input file in the same
directory.
In general an SCF run in Fleur follows the flow chart sketched in figure 1. After entering the SCF loop the current density is used to construct an effective potential for which the Kohn-Sham equations are solved. From the resulting eigenfunctions a new density is generated and it is checked whether it is similar enough to the input density auch that a self-consistent solution is found. If this is not the case a new density has to be constructed from all densities generated up to this point and another SCF iteration is started with this new density. If a self-sonsistent solution is found the program exits the loop.
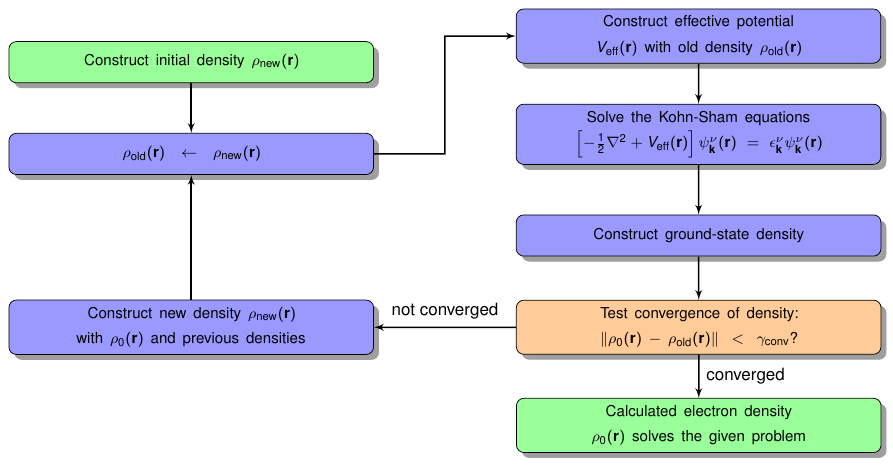
The processing of the SCF loop is directly affected by several parameters in the input file. The most important parameters determine when the program comes to a controlled end. For this one can define the maximal number of iterations to be performed and also a convergence criterion that leads to an immediate stop.
In the Fleur input file the maximum number of iterations to be performed is specified
in calculationSetup/scfLoop/@itmax. The convergence criterion defining the allowed distance
between the input and output densities for a converged solution is specified in calculationSetup/scfLoop/@minDistance.
By default a distance of less than is considered to indicate a converged calculation.
Slightly larger distances are also often tolerable.
Note that the presence of these two halting criteria implies that a finished Fleur run does not necessarily result in a self-sonsistent density. One should always check the distances between the densities for the last performed iteration.
The distances for each iteration of the current Fleur run is written out to the out file as
well as to the out.xml file. One can easily see the distances from all iterations by
executing grep distance out.
Beyond the halting criteria the configuration of the density mixing scheme plays an important role for SCF calculations. This is set by specifying the general type of the mixing procedure on the one hand and by parametrizing the mixing procedure on the other hand. For the parametrization a single mixing parameter has to be specified.
The type of mixing scheme is specified in calculationSetup/scfLoop/imix. The Anderson mixing typically
is a very efficient choice. If reaching convergence is difficult it is sometimes reasonable to perform parts of the
calculation with straight mixing. The mixing parameter is set in calculationSetup/scfLoop/alpha.
Several mixing schemes construct the next density on the basis of all densities calculated up to some point. Since the model assumptions for these mixing schemes might not be perfectly fulfilled it may be a good idea to delete this history of previous densities from time to time.
In the input file the parameter calculationSetup/scfLoop/maxIterBroyd specifies after how many SCF iterations
the mixing history is reset. Furthermore whenever a Fleur SCF run halts without obtaining a self-consistent
density a file mixing_history with the mixing history up to this point is written out. It is read in again
if Fleur is started in the same directory. By deleting it Fleur starts with an empty mixing history.
For metallic materials the Kerker preconditioner might speed up SCF convergence. Usage of the preconditioner becomes the more important the larger the system's unit cell or film thickness is. In Fleur it is currently available for non-magnetic and collinear-magnetic structures.
To switch preconditioning on, the optional preconditioning parameter calculationSetup/scfLoop/precondParam has
to be set to a positive value. The optimal parameter is material-dependent and is about 0.8 for most metals.